A foreign medical device manufacturer who wishes to be an approval holder for its product in Japan can leverage Designated Marketing Authorization Holder (DMAH) system.
Cobridge’s DMAH licenses allow it to handle medical devices (Class II through Class IV).
You can check the business model using DMAH services from the “DMAH Services Proposal Video” below.
The DMAH System in Japan
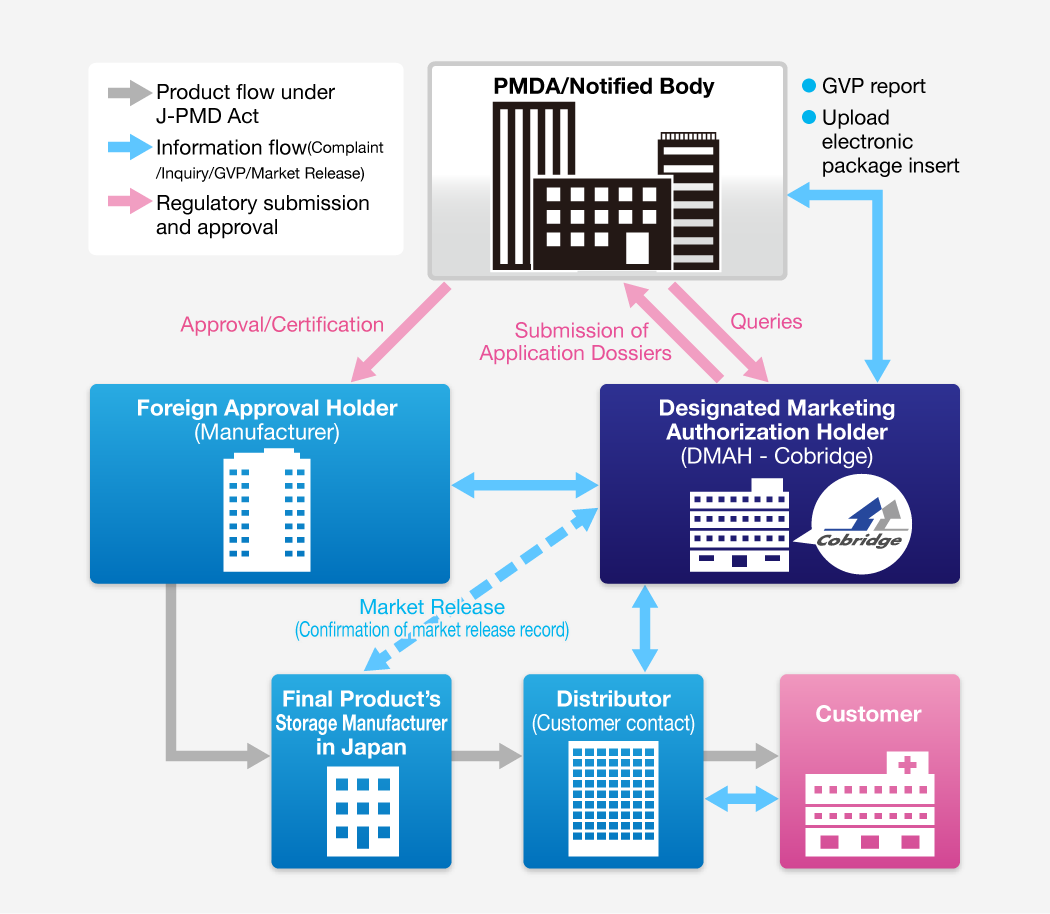
With the Japanese Pharmaceutical Affairs Law (PAL) amendments, which came into full force in 2005, foreign medical product companies could freely choose an DMAH to apply products’ registration as an applicant and own the approvals.
Foreign medical products companies need to perform the necessary regulatory-compliance obligations for the imported product as an product’s approval holder. But, most important, the DMAH need not be the Japanese distributor, thus product’s approval holder can freely choose their distributors.
This has important implications for overseas medical products producers wishing to export to the Japanese market.
Multifaceted Roles of the DMAH
Post-approval activities of the DMAH fall into two categories, Quality Management System (QMS) for medical devices and Good Vigilance Practice (GVP), both of which are defined in the PAL and associated regulations (ministerial ordinances). QMS relates to the monitoring of product quality, including batch release in Japan and any product complaints from the field. GVP relates to pharmacovigilance activities. Thus, the gamut of post-approval activities performed by Cobridge in its role as DMAH include (a) serving quality assurance duties for product release in Japan upon execution of QMS agreement with the Japanese importer licensed as a labeling, packaging and storage manufacturer, (b) performing post-marketing safety surveillance duties for the product in Japan upon execution of GVP agreements with the Japanese importer-distributor, (c) serving as the PMDA contact concerning the product, (d) monitoring gazetted notifications and pronouncements from the MHLW that relate to the product, (e) collecting and relaying legally required information regarding the overseas medical products firm (changes in executive management, changes in particulars regarding the manufacturing plant, etc.) to the PMDA, (f) coordinating the reporting of foreign safety reports to the PMDA and MHLW, and (g) preparing and filing initial and follow-up safety reports, and annual reports to the MHLW for adverse events that occur in Japan.